library(ggplot2)
library(ggseqlogo)5 Sequence Logo
5.1 Introduction
Sequence logos are a graphical representation of the sequence conservation in a multiple sequence alignment. They provide a way to visualize the relative frequencies of nucleotides or amino acids at each position in the alignment. The height of each letter in the logo corresponds to the frequency of the residue at that position. In R, the ggseqlogo package provides a set of functions to create sequence logos using the ggplot2 framework. To install this package, run install.packages("ggseqlogo"). Once installed, you can load the package using library(ggseqlogo).
5.2 Making a sequence logo
To make a sequence logo, you need to provide a vector of sequences. The sequences can be in the form of a character vector or a named list of sequences. The ggseqlogo function takes the sequences as input and generates the logo.
In the code below, we’ll first create a character vector of four tetra-nucleotide sequences.
my_align1 <- c("ATGC","ATTC","AGGC","ATAT")The ggseqlogo function creates a sequence logo from these sequences. The method keyword argument specifies the method used to calculate the height of each letter in the logo. The default method is “bits”, which uses Shannon entropy to calculate the height of each letter. Other methods include “prob” (probability).
ggseqlogo(my_align1, method="prob")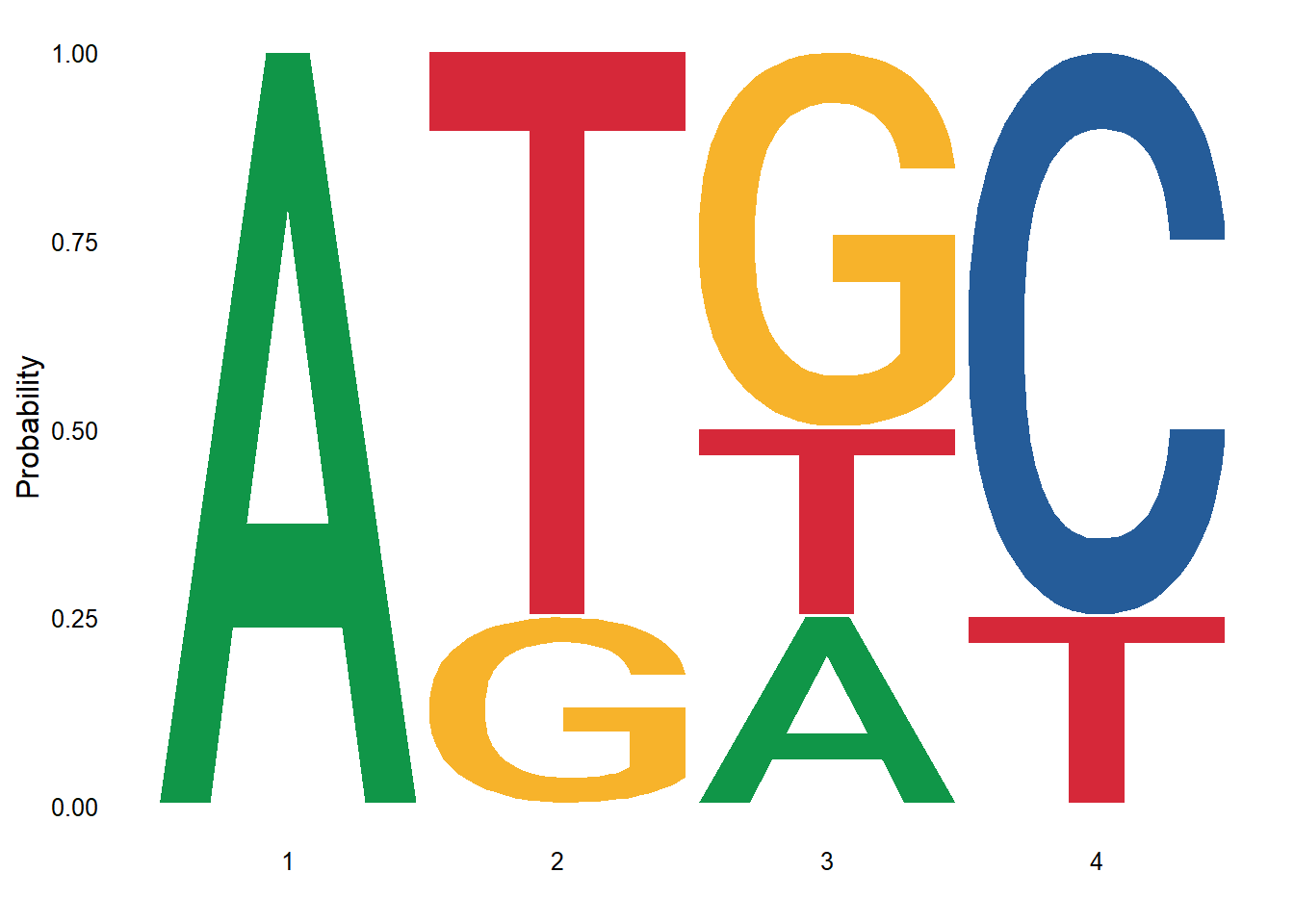
The ggseqlogo package has three sample datasets and can be loaded using the data function. The datasets - seqs_aa, and seqs_dna have list of 4 and 12 sequences respectively. The individual sequences in these lists can be accessed using the $ operator, e.g. seqs_aa$AKT1.
data(ggseqlogo_sample)
str(seqs_aa)List of 4
$ AKT1 : chr [1:172] "VVGARRSSWRVVSSI" "GPRSRSRSRDRRRKE" "LLCLRRSSLKAYGNG" "TERPRPNTFIIRCLQ" ...
$ CDK2 : chr [1:444] "LGPYEAVTPLTKAAD" "SGSESGYTTPKKRKA" "GSESGYTTPKKRKAR" "VTTQTPLTPEQLRAV" ...
$ AURKB : chr [1:106] "ITVTRRVTAYTVDVT" "PWPYGRQTAPSGLST" "APSLRRKTMCGTLDY" "PSKKRTQSIQGKGKG" ...
$ CSNK2A2: chr [1:80] "KHEEEEWTDDDLVES" "VWDHIEVSDDEDETH" "TSADVKMSSSEEVSW" "SADVKMSSSEEVSWI" ...str(seqs_dna)List of 12
$ MA0001.1: chr [1:97] "CCATATATAG" "CCATATATAG" "CCATAAATAG" "CCATAAATAG" ...
$ MA0002.1: chr [1:26] "AATTGTGGTTA" "ATCTGTGGTTA" "AATTGTGGTAA" "TTCTGCGGTTA" ...
$ MA0004.1: chr [1:20] "CACGTG" "CACGTG" "CACGTG" "CACGTG" ...
$ MA0005.1: chr [1:90] "CCTAATTGGGC" "CCTAATTTGGC" "CCTAATCGGGC" "CCTAATCGGGC" ...
$ MA0006.1: chr [1:24] "CGCGTG" "CGCGTG" "CGCGTG" "CGCGTG" ...
$ MA0007.1: chr [1:24] "AAAAGTACACCCTGTACCGACA" "CTAAGCACACCGTGTCCCAGTC" "TTAAGAACACTCTGTACGACAC" "AGTAGAACATAATGTTCCGACA" ...
$ MA0008.1: chr [1:25] "CAATTATT" "CAATTATT" "CAATTATT" "CAATTATT" ...
$ MA0009.1: chr [1:40] "CTAGGTGTGAA" "CTAGGTGTGAA" "CTAGGTGTGAA" "CTAGGTGTGAA" ...
$ MA0010.1: chr [1:9] "CTAATTGGCAAATG" "ATAATAAACAAAAC" "GACATAGACAAGAC" "GTCTTTCACAAATA" ...
$ MA0011.1: chr [1:12] "AACTATTT" "TGCTAGTT" "TCCTAGTT" "TTCTATTC" ...
$ MA0012.1: chr [1:12] "TAAACTTGTTG" "TAAACTAAAGC" "TCAACTAGGAT" "TAAACAAAACC" ...
$ MA0013.1: chr [1:6] "TTGTGAAAGAC" "AAGTAAACTAA" "TAATAAACAAA" "TAATAAACAAA" ...Below is the sequence logo for seqs_dna$MA0001.1. The optional font keyword argument is used to specify the font for the logo.
ggseqlogo(seqs_dna$MA0001.1, font="helvetica_light") 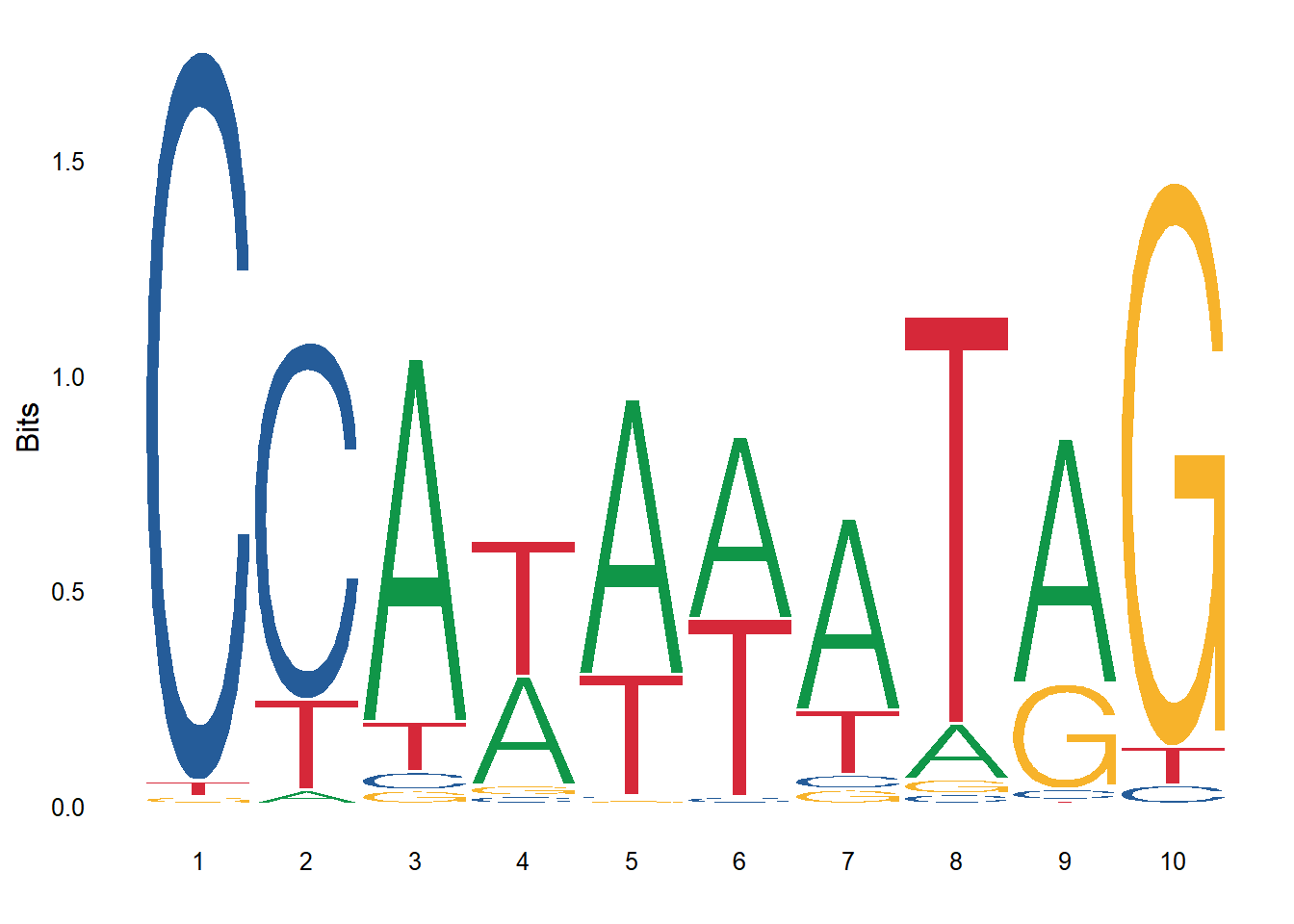
Similarly, we can add custom annotation to the logo. For example, the logo below has a rectangle within the logo to highlight a specific set of residue positions.
ggseqlogo(seqs_dna$MA0001.1, font="roboto_slab_light") +
annotate('rect', xmin = 3.4, xmax = 7.6, ymin = -0.05, ymax = 1, alpha = .1, col='blue', fill='yellow', linewidth=2)
5.3 Using alignment file
We can also read a multiple sequence alignment and render a logo from it. The read.alignment function from the seqinr package is used to read the alignment file. Subsequently, the desired region of the alignment is extracted using the substr function. Also, note that the toupper function is used to convert the sequences to uppercase. Finally, the ggseqlogo function is then used to create the logo from the extracted region. The alignment file used in the code below is available here.
library(seqinr)
prot_align <- read.alignment("DEAD2.aln", format = "clustal")
align_motif <- substr(toupper(prot_align$seq), 443, 448)
ggseqlogo(align_motif, seq_type="aa", method='prob')
We can render the logo using bits or prob as method. The logo can be saved as any other ggplot using the ggsave function.
library(patchwork)
p1 <- ggseqlogo(align_motif, seq_type="aa")
p2 <- ggseqlogo(align_motif, seq_type="aa", method="prob")
p1+p2+ plot_layout(guides = "collect") & theme(legend.position = "bottom")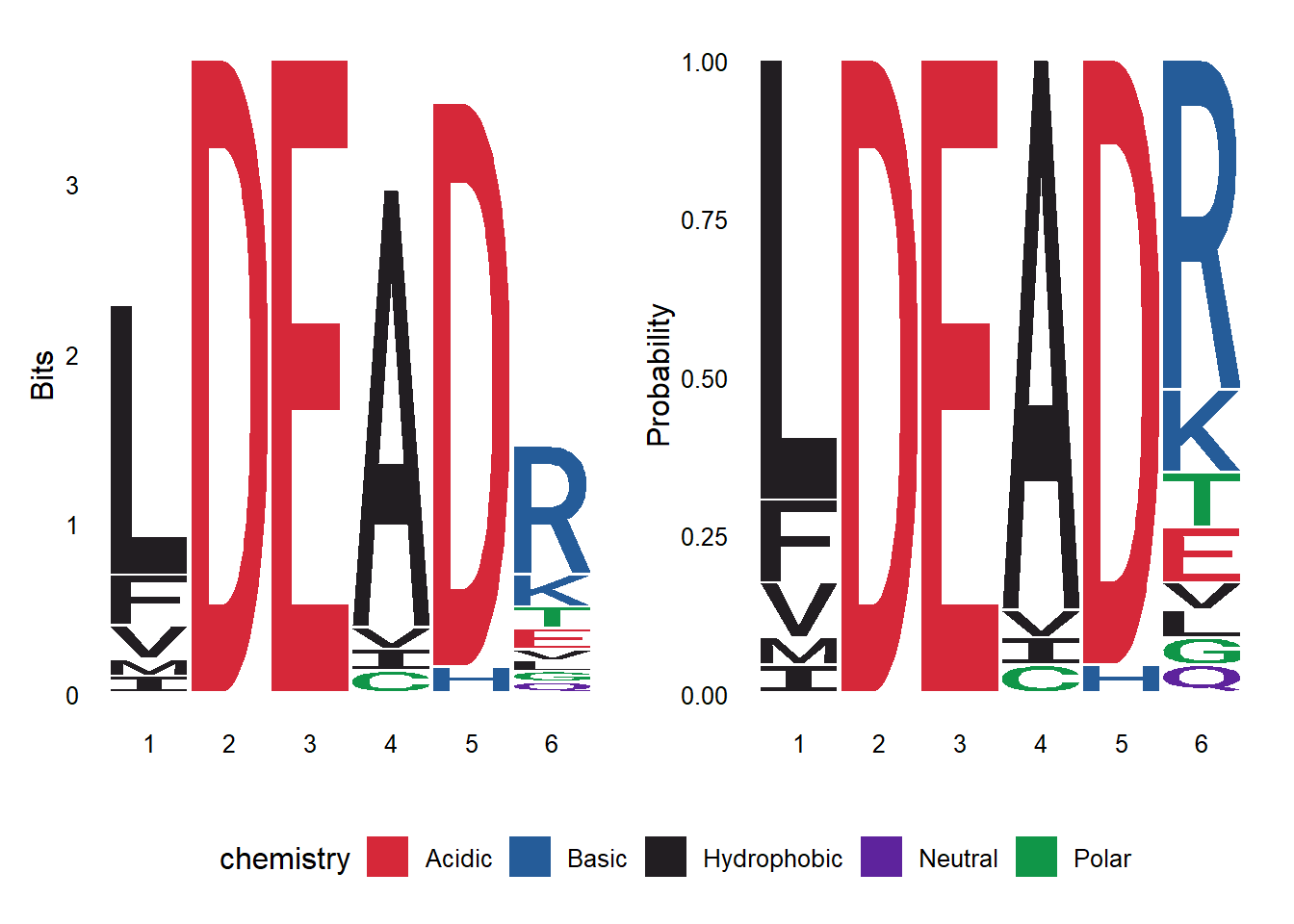
#ggsave("seqlogo_2.png", width = 6, height = 3, units = "in", dpi = 300)